CMI researchers at Ames National Laboratory conducted the research for this highlight
Innovation
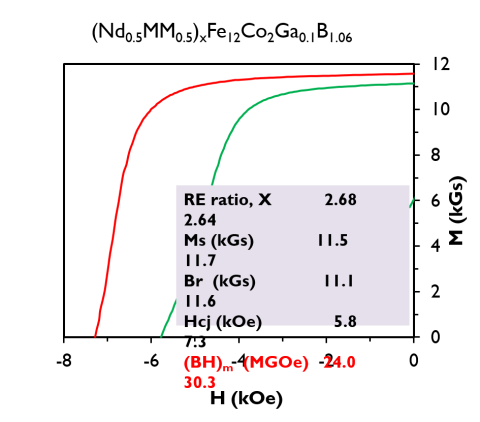
An NdFeB-based magnet with 40% of the Nd replaced by abundant rare earths La and Ce achieved an energy product of 30 MG-Oe.
Achievement
Performance approaching that of entry-level grades of NdFeB magnets at reduced cost and effective criticality.
Significance and Impact
Simultaneous usage of La and Ce may enable substantial effective criticality reduction. Usage of Mountain Pass mischmetal component convenient but innovation is performance at reduced Nd usage.
Hub Goal Addressed
Industrial adoption of CMI Technologies.