
A collaboration between ORNL and UC-Davis led to this highlight
Achievement
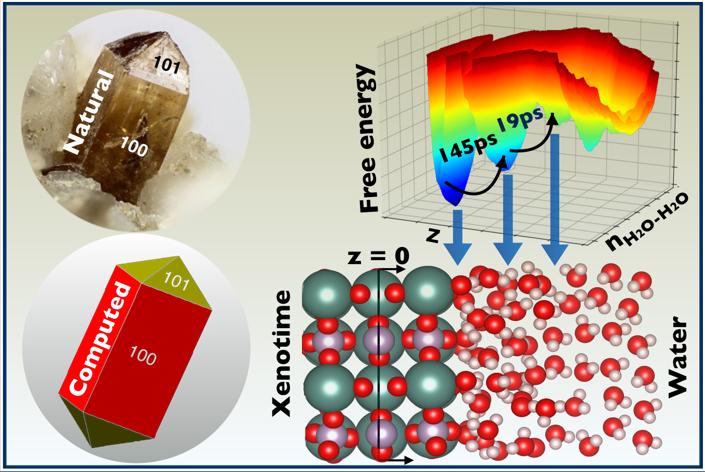
Ab initio molecular dynamics (AIMD) simulations resolve the hydration structure and water-exchange kinetics at the water–xenotime interface.
Significance and impact
AIMD simulations establish that {100} xenotime surface can only accommodate monodentate coordination of a bidentate hydroxamate collector. Differences in the hydration structure and water-exchange kinetics imposed by different ligands could be used to develop design strategies to enhance flotation selectivity of RE minerals.
Details and next steps
- Different interfacial (Y3+ and PO_4^(3−)-bound) and bulk-like water layers with distinct orientations are identified on the 2D-free energy surface.
- A Marcus rate theory reveals that water exchange occurs on 10s to 100s of ps timescales, exhibiting strong sensitivity to ligand orientation, which prompts us to develop new strategies for selective flotation of RE.
- Our methodology is transferable to the study of water-exchange kinetics and ligand adsorption on bastnaesite.