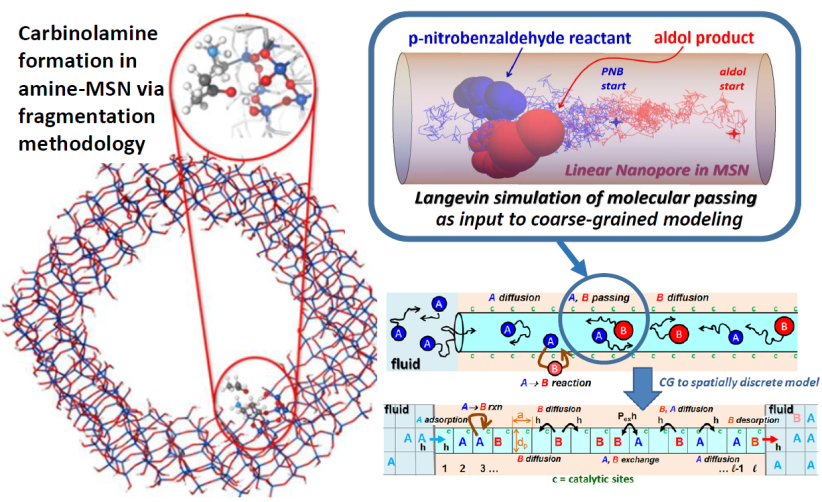
in functionalized mesoporous silicananoparticles (MSN). This includes detailed quantum chemical analysis of reaction mechanisms, Langevin simulation of key diffusive dynamics, coarse-grained modeling of the overall reaction process and kinetics.
This project supports efforts in electronic structure theory and non-equilibrium statistical mechanical & multiscale modeling of complex catalytic chemical systems. The primary focus is on the development and application of methods that enable the study of condensed-phase chemistry and surface reaction phenomena, especially heterogeneous catalysis. Also of interest are solvent effects and homogeneous organometallic catalysis. Electronic structure theory efforts integrate development of fundamental theory, expanding the capability for accurate treatment of large systems of interest to USDOE BES, with optimal strategies for computational implementation within GAMESS. Theory development includes fragment molecular orbital, effective fragment molecular orbital and effective fragment potential approaches with applications to liquid-solid interfaces, and embedding methods for solid surfaces. Statistical mechanical and multiscale modeling studies also focus on catalytic reaction-diffusion phenomena and catalytic nanomaterials. This modeling often incorporates input from relevant electronic structure analyses. A core focus in this effort is on molecular-level and coarse-grained modeling the interplay between restricted transport and catalytic reaction in functionalized nanoporous materials. Another major effort is on the predictive molecular-level modeling of chemisorption and heterogeneous catalysis on metal surfaces and nanoclusters. In addition, our modeling explores the synthesis and stability of catalytic nanomaterials, currently focusing on metallic nanocrystals formed in the solution-phase or by deposition.
This research is supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences through the Ames Laboratory. The Ames Laboratory is operated for the U.S. Department of Energy by Iowa State University under Contract No. DEAC02-07CH11358.
Principal Investigator: James Evans
Co-PIs: Mark Gordon, Klaus Ruedenberg, Theresa Windus
Staff Scientists: Da-Jiang Liu, Georg Schoendorff, Yong Han, Federico Zahariev
Post Doctoral Researcher: Andres Garcia
News & Highlights
 Research Highlight
Research Highlight
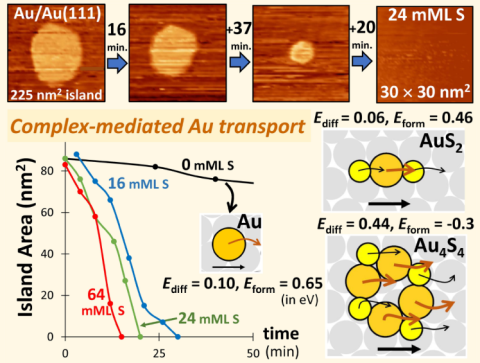
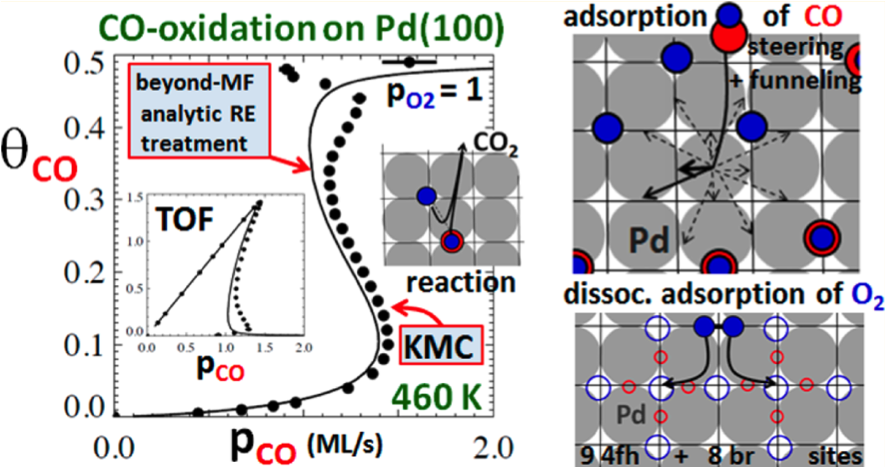
Enhanced dynamics of nanostructures on Au(111) surfaces: facile mass transport mediated by adsorbed Au-S complexes
 Research Highlight
Research Highlight
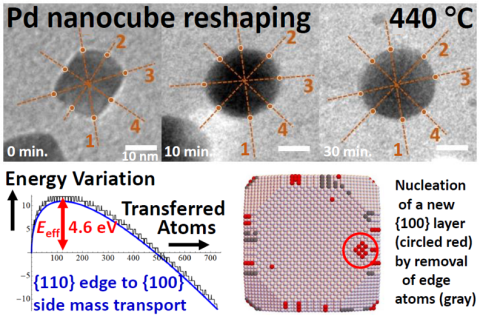
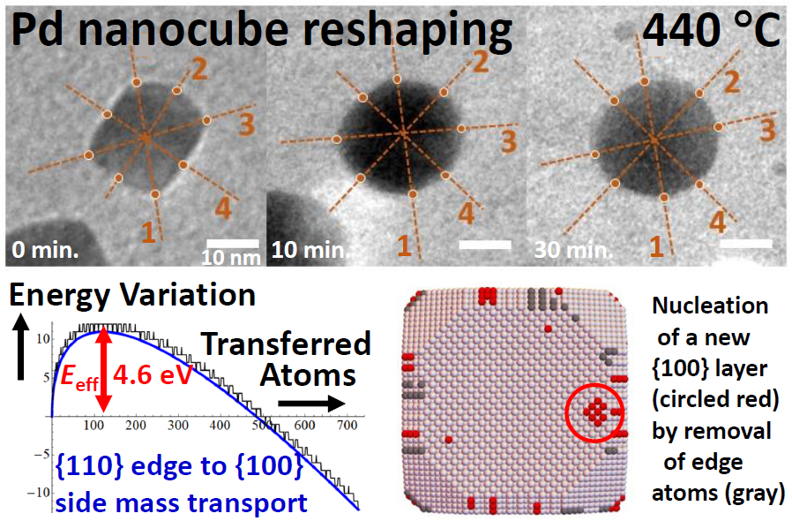
Reshaping of Truncated Pd Nanocubes
 Research Highlight
Research Highlight
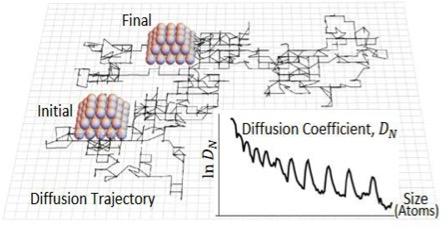
Complex oscillatory decrease with size in the diffusivity of epitaxially-supported 3D metal nanoclusters
 Research Highlight
Research Highlight
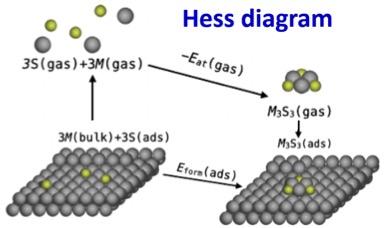
Stability of metal-sulfur complexes on metal surfaces and ramifications for the fluxional dynamics of surfaces under reaction conditions
 Research Highlight
Research Highlight