Overview
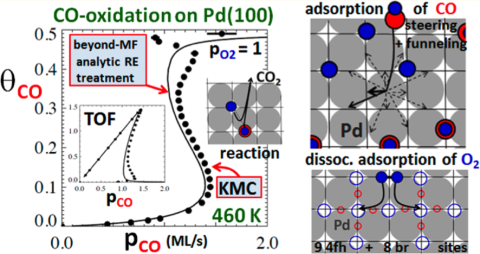
A long-standing challenge is the development of realistic molecular-level models for catalytic reactions on metal surfaces under low-pressure conditions. Here interactions between adsorbed reactant species produce subtle reactant adlayer ordering and also impact both (non-reactive) desorption and reaction kinetics. Also, there has been little attention paid to the cooperative nature of adsorption on surface covered by mixed reactant adlayers. Yet this is critical for reliable prediction of behavior under steady-state flow conditions (turn-over frequencies, bifurcations in steady=states, etc. which is rarely addressed in modeling.)
Traditional mean-field chemical kinetics has been immensely useful for analysis of such behavior under steady-state flow conditions. Yet it fails to account for such basic features as reactant adlayer ordering. This deficiency is corrected in the current work adapting tools from statistical mechanics to incorporate the effect of adlayer ordering and of complex cooperative adsorption dynamics into a reliable treatments of chemical kinetics.
Another fundamental challenge for this modeling is the sensitivity of kinetics on key energetics (e.g., adsorption energies and related desorption barriers, as well as reaction barriers) for the system. The limitations of DFT formalisms for prediction of such energies are well recognized. For CO-oxidation on Pd(100), the CO desorption barrier is found to control the regime of bistability (coexistence of a reactive steady-state with low CO-coverage and a near-CO-poisoned state). Extensive high-level analysis of this barrier is provided to correct DFT limitations.
Da-Jiang Liu, Federico Zahariev, Mark S. Gordon and James W. Evans, Predictive Beyond-Mean-Field Rate Equations for Multisite Lattice− Gas Models of Catalytic Surface Reactions: CO Oxidation on Pd(100). J. Physical Chemistry C, 2016, 120, 28639-28653.