Thrust 1 - AI/ML-assisted materials discovery and design framework
Li (Co-leader), Wang (Co-leader), Xia, Moraru, Ye, Yao, Antropov, Richard
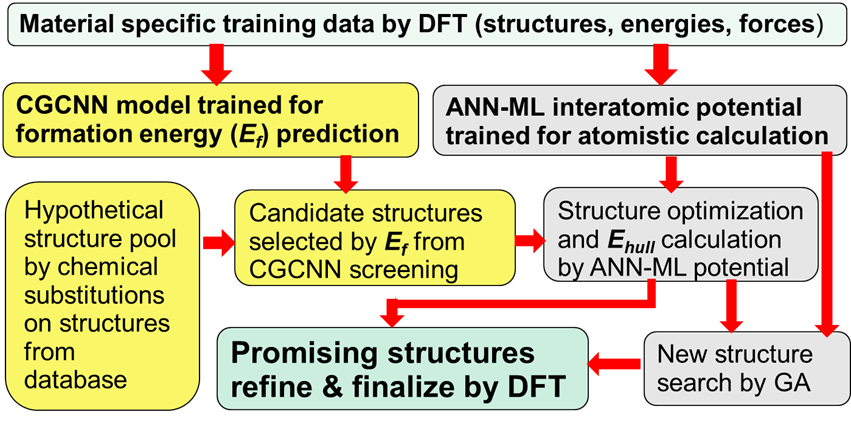
This thrust develops an AI/ML-assisted framework on high-performance and exascale computers to significantly accelerate discovery and design for functional materials with a focus on emergent magnetic and superconducting compounds. Our framework integrates AI/ML tools such as crystal graph convolutional neural network (CGCNN), artificial neural network (ANN) ML interatomic potentials, and materials databases, with state-of-the-art computational methods including adaptive genetic algorithm (AGA) and first-principles calculations on high-performance and exascale computers. Our implementation of the AI/ML-assisted framework leverages recent GPU-based ANNs and first-principles calculation codes and a parallel, task-based workflow manager Parsl. This approach significantly reduces the time to solution for materials discovery and design through
Research Progress
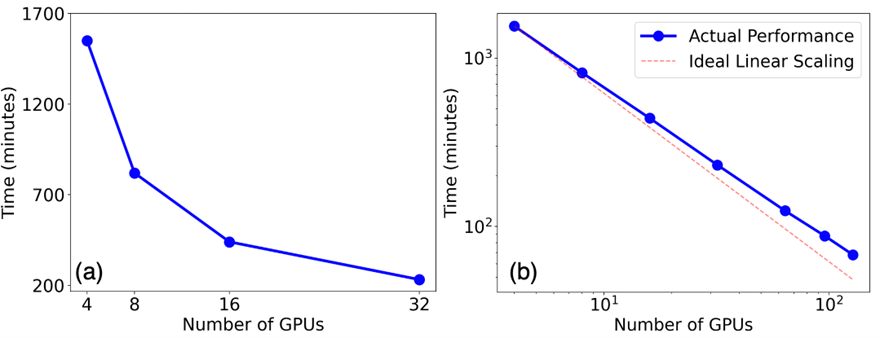
The framework is ready for applications to search for ternary compounds. The speedup of the framework scales almost linearly with the number of GPUs. For any ternary system with three chemical elements, it produces a map of comprehensive composition-structure-energy landscape in just a few hours. Fig. 1 shows the scaling performance for Na-B-C ternary compounds. About 1000 candidate structures were selected from CGCNN for first-principles calculations. Using 32 GPUs on Perlmutter at NERSC, the energy calculations completed in about 3-4 hours as shown in Fig. 1 (a). The almost linear strong scaling performance can also be seen from Fig. 1 (b). For most ternary systems, about 2000 candidate structures per system will be selected for DFT calculations, which will complete within a few hours using 64 GPUs. Another notable advantage of the framework is its capability to perform the search for an arbitrary number of ternary systems simultaneously, and the parallel computation can scale up to several thousand GPUs. This means the ML framework can predict stable and metastable compounds for 100 different ternary systems in a few hours if 6000 GPUs on Perlmutter at NERSC are available.
Thrust 2 - Computational prediction of synthesis pathways
(Zhang (Leader), Li, Xia, Ye, Moraru, Wang, Yao, Richard)
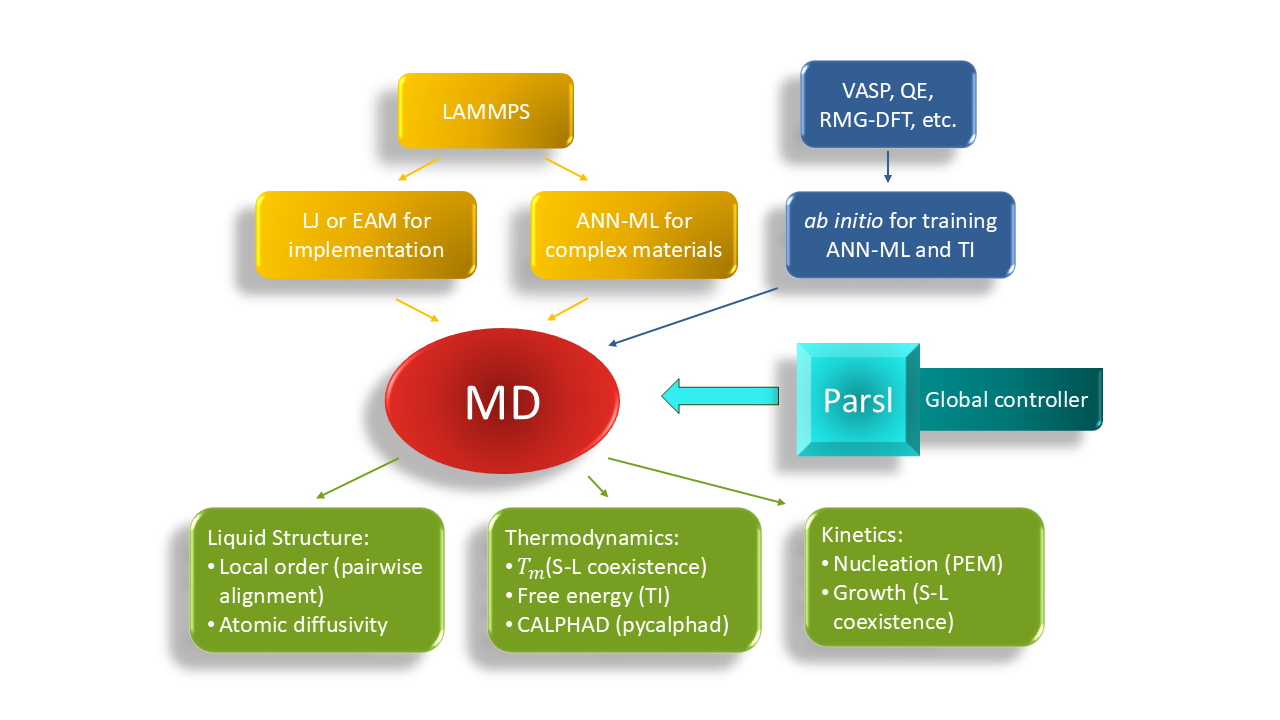
This thrust develops and executes exascale workflows to efficiently acquire thermodynamics data and phase selection/nucleation kinetics. The goals are to understand the local energy landscape between transient or intermediate products, and to guide precision synthesis by suggesting synthesis pathways. Currently, many computationally predicted new compounds cannot be synthesized, largely due to a lack of knowledge about the necessary synthetic pathways. We leverage molecular dynamics (MD) simulation package LAMMPS, an Exascale Computing Project (ECP) code supported by DOE, and incorporate several robust and powerful computational methods and codes developed at Ames National Laboratory (Ames Lab) to substantially speed up the prediction of synthesis pathways for complex functional materials. Accurate and efficient ANN-ML interatomic potentials are developed to ensure reliable and efficient MD simulations of complex materials.
Research Progress
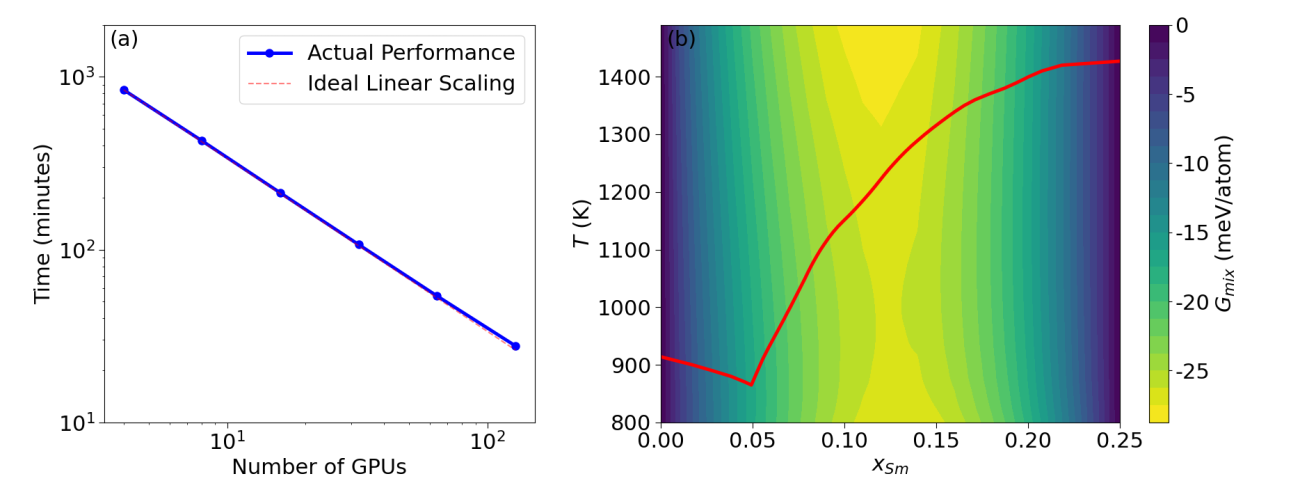
The phase diagram based on precise free energy calculations provides essential guidance for materials synthesis. The thermodynamic integration (TI) technique is highly accurate for free energy calculations, yet it is also resource-demanding. For instance, hundreds of MD jobs are required to map out the free energy of a binary solution phase (such as liquid) as a function of temperature (T) and composition (x). We have set up a framework that uses Parsl, an open-source Python library for parallel programming, to efficiently manage the MD jobs according to the available resources. Fig. 2 (a) shows almost ideal strong scaling of this framework for the calculation of the free energy of the Al-Sm liquid in the Al-rich regime (x_Sm≤0.25) based on an EAM potential. Fig. 2 (b) gives the resulting mixing Gibbs free energy G_mix (x,T) of Al-Sm liquid referenced to the two end compositions: pure Al liquid and Al0.75Sm0.25 liquid. The free energy of Al-Sm liquids and the two competing solid phases fcc-Al and Al3Sm can be used in CALPHAD modeling to obtain the melting line shown as the red curve in Fig. 2(b).