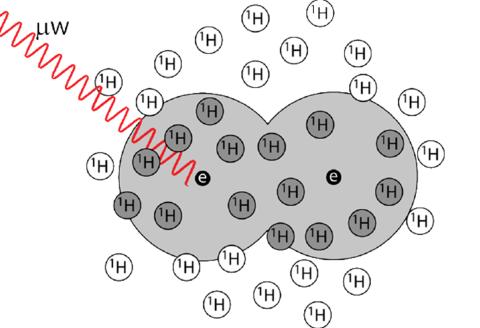
Dynamic Nuclear Polarization (DNP) is a powerful emerging technique capable of improving the sensitivity of solid-state NMR by 2-4 orders of magnitude and lead to the atomistic description of surfaces and interfaces. Additional improvements can be achieved by increasing the performance of the radical polarizing agent combined with appropriate sample preparation strategies. Accelerating the discovery of improved polarizing agents through simulation requires a proper treatment of the concerted dynamics of thousands of spins over several seconds, far exceeding conventional approaches incorporating only a handful of spins. This was achieved by restricted state space methods and a Monte Carlo optimization algorithm to calculate the enhancements directly, without worrying about the timescale of the interactions. This new approach enabled the calculation of DNP enhancements in systems containing upwards of 50 spins allowing for the importance of the spin diffusion and the diffusion barrier to be determined. Calculations demonstrated that high radical concentrations are required forfast-MAS. Large scale simulations of partially-deuterated biradicals reproduced experimental trend sin DNP enhancements while smaller spin systems could not. These developments open the door to the fully ab initio simulation of real solutions and the quantitative reproduction of experimental DNP enhancement factors.
F. A. Perras, M. Pruski.* Large-scale ab initio simulations of MAS DNP enhancements using a Monte Carlo optimization strategy, Journal of Chemical Physics, 2018, 149, 154202. DOI: 10.1063/1.5042651