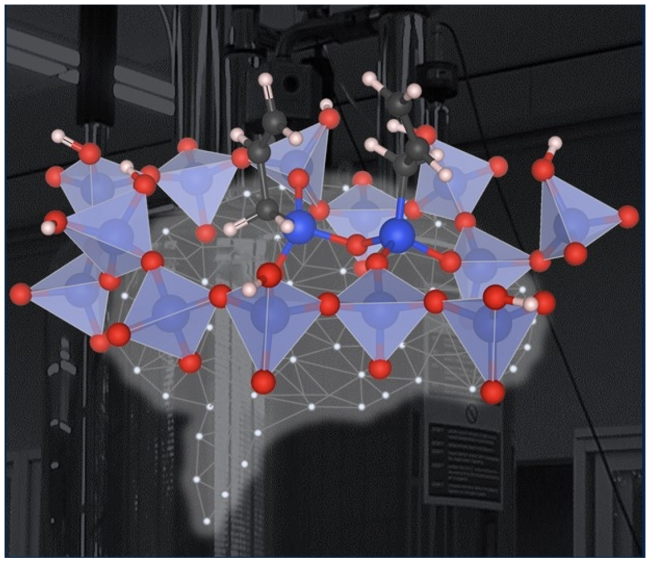
Scientific Achievement
A method was established to study the geometry and arrangement of dynamic surface species.
Significance and Impact
Molecular motions are predicted by Molecular Dynamics (MD) simulations and taken into account in solid state nuclear magnetic resonance (NMR) structural analysis of surface species. Detailed models of the arrangement of sites on the catalyst surfaces will contribute to their design and computational modeling.
Research Details
- The effect of molecular motion on NMR parameters is predicted from MD simulated atom trajectories.
- A machine learning approach is used to generate the relatively long simulation times required to estimate motionally averaged NMR parameters.
- Theory and experiment identify specific short contacts between surface species.
Kobayashi, T.*; Liu, D.-J.*; Perras, F.A. Chem. Commun. 58, 13939 (2022). DOI: https://doi.org/10.1039/d2cc05861h