CMI scientists at Ames Laboratory conducted this research.
Achievement:
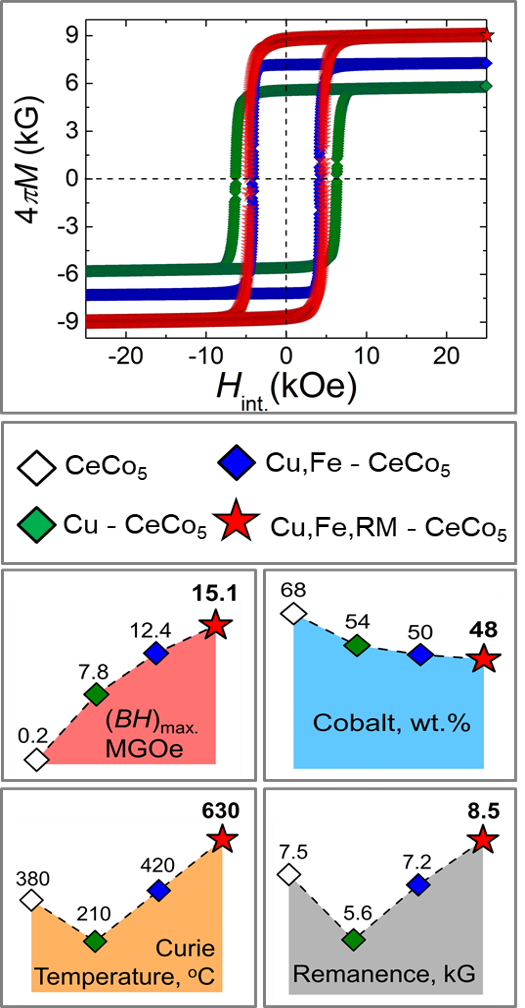
- Polycrystalline Cu, Fe and RM-doped CeCo5 systems (RM – refractory metal) with energy performances within (BH)max. = 13.0 – 15.5 MGOe
- Uses up to 30 % less Cobalt than pure CeCo5
- Ce-based magnet with thermal characteristics approaching the levels of Sm – Co magnet grades, i.e., 600 – 650 oC
Significance and Impact:
Co-lean Ce-based gap magnets that are based on more abundant material, versatile, process effective, and can be used in wide range of applications associated with automotive and sensor industries
Details and Next Steps:
- Leveraging team’s unique “composition-temperature-time” expertise, Co-lean, high-temperature Phase I 1:5-type, Ce-based gap magnets with energy performance beyond 15 MGOe could be synthesized
- Next step is to improve understanding of the coercivity mechanism for these versatile material systems